

Vielversprechende Ergebnisse in der Entwicklung neuer Arzneimittel durch ML
Traditionell ist die Entwicklung neuer Medikamente von Natur aus komplex. Der Prozess der Wirkstoffentdeckung beginnt mit der Identifizierung der Krankheitsursache, gefolgt von der Erstellung einer Liste von Wirkstoffkandidaten und deren anschließender Überprüfung – langwierig und kostenintensiv.
Dank jüngster Fortschritte im maschinellen Lernen (ML) und einer größeren Verfügbarkeit an Daten sind wir nun in der Lage, diese komplexen Probleme datengesteuert zu lösen. Im folgenden Artikel erfahren Sie, welche Auswirkungen fortschrittlicher ML-Methoden und Partnerschaften auf die Beschleunigung der Wirkstoffentdeckung haben können.
Screening- oder generative Strategien
Im Bereich des maschinellen Lernens gehen Forschungsgruppen molekulare Designherausforderungen in der Regel mit einer von zwei Strategien an:
- Screening: Überprüfung einer Liste von Molekülen und ihrer Eigenschaften. ML-Modelle werden trainiert, um Struktur-Eigenschafts-Beziehungen herzustellen.
- Generativ: Iteratives Erstellen neuer Moleküldesigns unter Verwendung von Feedback aus prädiktiven Modellen aus einem Screening-Szenario, um die Qualität der Kandidaten zu beurteilen.
Bei beiden Methoden werde Zieleigenschaften erforscht und potenzielle Kandidaten zur Verwendung in Medikamenten identifiziert. Jedoch kann der Mangel an Daten zu bestimmten Eigenschaften kann die Robustheit der prädiktiven Modelle beeinträchtigen. Dabei können ML-Techniken helfen, neuartige Verbindungen zu identifizieren, insbesondere wenn nur wenige Daten verfügbar sind.
Unsere Hypothese ist, dass Verbindungen mit ähnlichen Strukturen tendenziell ähnliche Funktionen haben. Wir haben dies geprüft, indem wir neuartige Koagulans-Kandidaten identifiziert haben, die bei der Kontrolle der Blutgerinnung helfen.
Verbesserung der Behandlung von Blutgerinnseln mit Autoencodern
Neben der technischen Herausforderung ist die Kontrolle der Blutgerinnung wichtig für die Behandlung von Patienten nach Operationen und bei Gerinnungsstörungen (Antikoagulations-/Koagulationskaskaden) wie Thrombose oder Hämophilie.
Die Behandlung variiert je nach Fall. Man kann fehlende Proteine hinzufügen (Substitutionstherapie) oder das (Anti-)Koagulationsgleichgewicht korrigieren. Dies könnte bedeuten, bestimmte Proteine zu hemmen oder ihre Produktion zu blockieren. Zum Beispiel verstärkt die Unterdrückung von Antikoagulans-Proteinen die Koagulationsaktivität. Das Gegenteil funktioniert ebenfalls.
Unser Vorgang zur Entdeckung neuer Koagulans-Kandidaten basiert auf Autoencoder – eine Art von KI, die lernt, Daten zu komprimieren und dann zu rekonstruieren. Diese Autoencoder helfen, jedem Molekül eigene Koordinaten in einem „chemischen Raum“ zuzuweisen und bekannte Koagulantien und Antikoagulantien zu analysieren, die in diesem Raum verteilt sind.
Autoencoder: Werden oft verwendet, um Muster zu finden oder Rauschen in Informationen zu reduzieren.
Protein C: Ein Protein im Blut, das übermäßige Gerinnung verhindert und so für einen reibungslosen Blutfluss sorgt.
Thrombin: Ein Enzym, das bei einer Schnittwunde oder Verletzung Die Blutgerinnung fördert und so die Blutung stoppt.
Datensätze erstellen und analysieren
Damit Autoencoder optimal funktionieren, benötigen Sie einen soliden Datensatz. Um den Hemmungsdatensatz zu erstellen, nutzten wir die öffentlich zugängliche BindingDB-Datenbank (Zugriff im Juni 2023). Diese Datenbank stellt Protein- und Ligandenstrukturen im SMILES-Format (Simplified Molecular Input Line Entry System) zur Verfügung. Sie zeigt auch, wie stark Protein und Ligand interagieren.
Wir haben zwei Arten von Daten aus BindingDB extrahiert:
- Aktivitätsklasse (aktiv, inaktiv)
- Aktivitätsmaß (Hemmkonstante Ki)
Wir konzentrierten uns auf Thrombin und Protein C, die die Blutgerinnung kontrollieren. Diese beiden gehören jeweils zu den Koagulations- und Antikoagulationskaskaden. Der Thrombin-Klassifikationsdatensatz umfasste 5.009 Liganden, von denen 3.323 basierend auf Ki, KdEC50 oder IC50 als aktiv eingestuft wurden. Der Thrombin-Regressionsdatensatz enthielt 2.270 Liganden mit verfügbaren Ki Werten, von denen 1.927 aktiv waren (Ki ≤ 10,000 nM).
Der Protein-C-Klassifikationsdatensatz enthielt 188 Protein-C-Liganden mit 103 aktiven. Für 125 Liganden ist der Ki Wert verfügbar, und 79 sind bestätigte aktive Protein-C-Inhibitoren. Die Anzahl der bekannten Protein-C-Inhibitoren war zu gering, um zuverlässige und robuste ML-Modelle zu trainieren. Zum Glück waren Moleküle mit bekannten Aktivitäten nicht erforderlich, um den Autoencoder zu trainieren. Das Ziel des Autoencoders ist es, die universellen chemischen Regeln der SMILES-Syntax zu lernen. Dieses Training verwendete große Sätze gültiger SMILES-Beispiele.
Neue Moleküle generieren
Mit einem detaillierten Datensatz begannen wir, neue Moleküle zu erzeugen. Unser Ansatz hat zwei Hauptphasen:
- Zuerst erstellten wir einen Autoencoder, um den chemischen Raum kleiner organischer Moleküle abzubilden.
- Dann generierten wir Kandidaten, indem wir diesen abgebildeten Raum erkundeten.
Autoencoder sind eine unüberwachte Methode, die darauf trainiert ist, Ausgaben mit Eingaben abzugleichen. Sie komprimieren zuerst die Eingangsinformationen in einen Engpass und dekomprimieren sie dann wieder zur Ausgabe. Jeder Datenpunkt hat seinen eigenen Einbettungsvektor, der als Koordinaten in einem Hyperraum fungiert. Dies gilt auch für Moleküle – diese können mit einzigartigen Koordinaten (einem Einbettungsvektor) in einem chemischen Hyperraum kodiert werden.
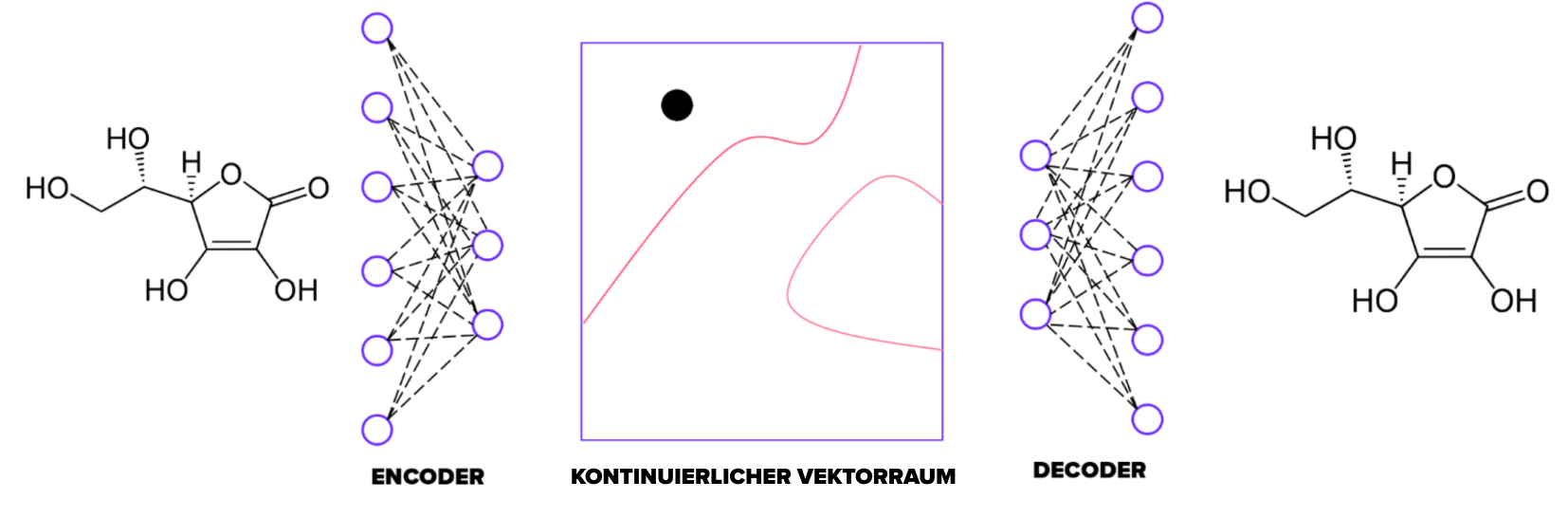
Der Autoencoder besteht aus zwei Teilen: einem Encoder und einem Decoder. Der Einbettungsvektor wird aus der Ausgabe des Encoders abgeleitet. In unserer Implementierung beträgt die Größe der Einbettung 100. Wir haben den Autoencoder durch Ändern der Anzahl der neuronalen Schichten, ihrer Typen, Formen und Aktivierungsfunktionen feinabgestimmt. Als Ergebnis hat der Encoder vier Faltungsschichten und eine vollständig verbundene Schicht. Der Decoder spiegelt diese Struktur mit einer vollständig verbundenen und vier Faltungsschichten wider.
- Thrombin-Affinitätsprädiktoren: Da die Datenverfügbarkeit für bekannte Thrombin-Inhibitoren hoch ist, haben wir entsprechende Prädiktoren trainiert und die Generierung von Antikoagulantien validiert. Wir verwendeten einen zweistufigen Bewertungsansatz, bei dem zuerst eine Hemmklasse (aktiv vs. nicht aktiv) vorhergesagt wurde und dann, falls aktiv, die Hemmkonstante (Ki) unter Verwendung von Ensembles verschiedener ML-Regressionsmodelle.
- Clusterung von Thrombin- und Protein-C-Inhibitoren: Im Einbettungsraum des Autoencoders identifizierte die Clusteranalyse mehrere Cluster für Protein-C- und Thrombin-Inhibitoren. Durch den Vergleich der charakteristischen Abstände innerhalb und zwischen diesen Clustern wählten wir einen „sicheren“ Radius von (dsep) = 0.8. Wie die Koordinaten im Einbettungsraum ist dieser Abstand dimensionslos. Der Radius soll bekannte Inhibitoren abdecken und gleichzeitig die Einbeziehung von Inhibitoren des entgegengesetzten Proteins vermeiden.
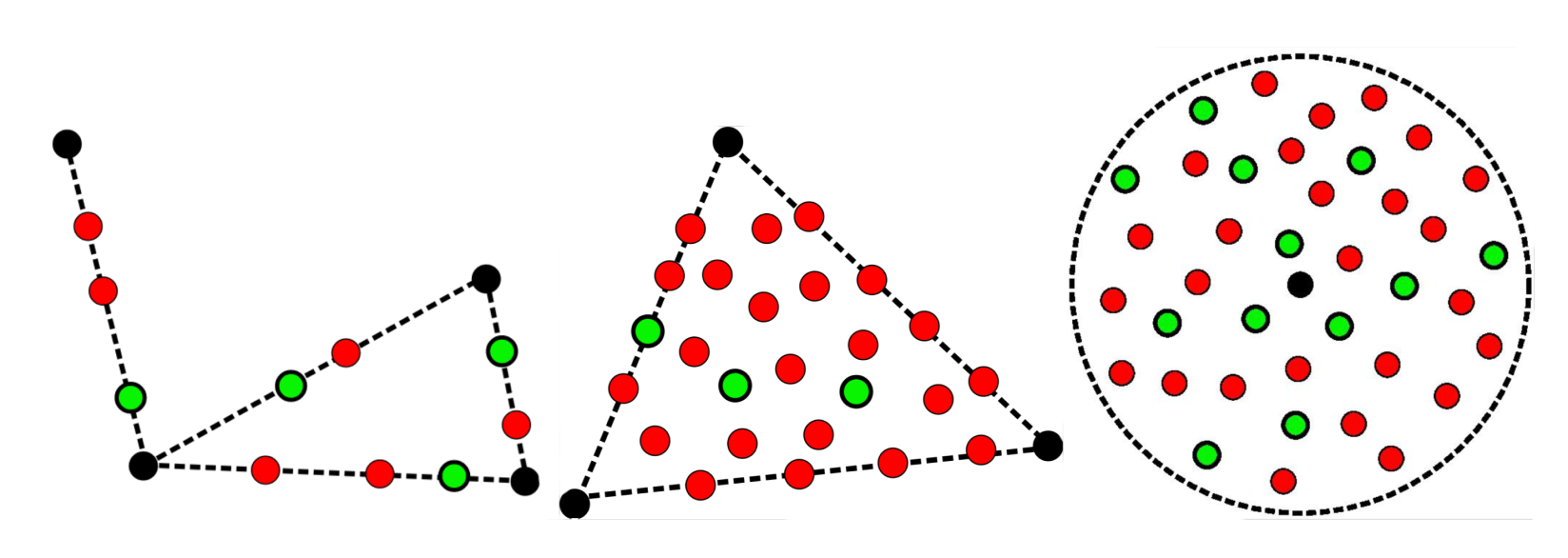
- Generierung neuer Molekülkandidaten: Verschiedene Strategien zur Generierung neuer Verbindungen wurden eingesetzt und geprüft. Wir interpolierten zwischen Paaren von Referenzverbindungen oder wählten Punkte innerhalb von dreieckähnlichen Domänen, die durch Tripletts von Verbindungen definiert sind. Um neue Kandidatenmoleküle in diesem Raum zu finden, zogen wir auch eine imaginäre Hypersphäre um eine Referenzverbindung. Alle generierten Einbettungen wurden:
- zu SMILES-Strings dekodiert.
- auf chemische Gültigkeit getestet.
- basierend auf der Nähe zu gewünschten oder unerwünschten Inhibitorgruppen gefiltert.
Gefüllte schwarze Kreise zeigen die bekannten Inhibitoren. Rote Kreise, die fehlerhafte SMILES darstellen, werden herausgefiltert. Die korrekten, in Grün, werden beibehalten und durchlaufen eine Reihe von Filtern, um sie zu bewerten und zu priorisieren.
MegaMolBART: Wir haben MegaMolBART, ein transformatorbasiertes Sprachmodell, neben unserem Autoencoder-basierten Ansatz einbezogen. Diese Lösung basiert auf der BART-Architektur (Bidirectional and Auto-Regressive Transformer). Es ist ein generatives KI-Modell (Gen AI), das durch eine Partnerschaft zwischen AstraZeneca und NVIDIA entwickelt wurde. MegaMolBART löst eine ähnliche Aufgabe wie unser Autoencoder-basierter Ansatz. Seine Schnittstelle ermöglicht die Generierung durch Interpolation oder durch Suche in der Nähe von Referenzverbindungen. Im Wesentlichen verwenden unser Ansatz und MegaMolBART dieselben generativen Techniken, haben aber einzigartige molekulare Einbettungen.
Generierte Protein-C-Thrombin-Inhibitor-Kandidaten: Wir verwendeten den Interpolationsansatz, um 200.000 Einbettungen zu generieren, was 5.736 korrekte SMILES ergab. Nachdem wir die ursprünglichen Thrombin-Inhibitoren entfernt hatten, blieben 5.354 einzigartige Moleküle übrig. Wir filterten nach Abständen zu den nächstgelegenen bekannten Thrombin-Inhibitoren und den nächstgelegenen starken Protein-C-Inhibitoren, und 3.150 einzigartige SMILES wurden akzeptiert, von denen 1.420 als aktiv vorhergesagt wurden.
Es wurden Hypersphären mit Radien bis zu einem „sicheren“ Abstand von dsep = 0.8 um jeden der 2.270 Thrombin-Liganden erstellt. Diese Methode ergab 34.957 einzigartige und chemisch korrekte Strukturen. Von diesen erfüllten 16.027 einzigartige SMILES unsere Akzeptanzkriterien basierend auf den zuvor genannten Abständen, wobei 6.369 als aktiv vorhergesagt wurden. Bei der Analyse der vorhergesagten log10 Ki-Werte für diese generierten Verbindungen im Vergleich zu den tatsächlichen log10 Ki-Werten von Referenzinhibitoren stellten wir fest, dass die vorhergesagten log10 Ki-Werte deutlich unter der Aktivitätsschwelle bleiben, wenn wir die Referenzinhibitoren mit log10 Ki unter zwei berücksichtigen.
Generierte Protein-C-Inhibitor-Kandidaten: Der Datensatz der Protein-C-Liganden enthielt 103 aktive Verbindungen. Wir verengten die Liste der Referenzinhibitoren auf solche mit einem log10 Ki unter zwei und wählten 15 starke Referenzinhibitoren aus. Mit diesen generierten wir 1.571 Protein-C-Inhibitor-Kandidaten mit den Hypersphären- und Interpolationsansätzen. Wir bewerteten die generierten Verbindungen auch nach ihren Synthetic Accessibility Scores (SAS), wobei der niedrigste SAS bei 3,86 aus der Hypersphärensuche stammte.
Die SAS-Werte für generierte Protein-C-Inhibitor-Kandidaten, die die Ghose-, Veber- oder Egan-Drug-Likeness-Filter passieren, sind größer als vier. Interessanterweise ist ein bestimmter Kandidat mit einem SAS von 6,28 allen generativen Techniken gemeinsam (siehe unten links).
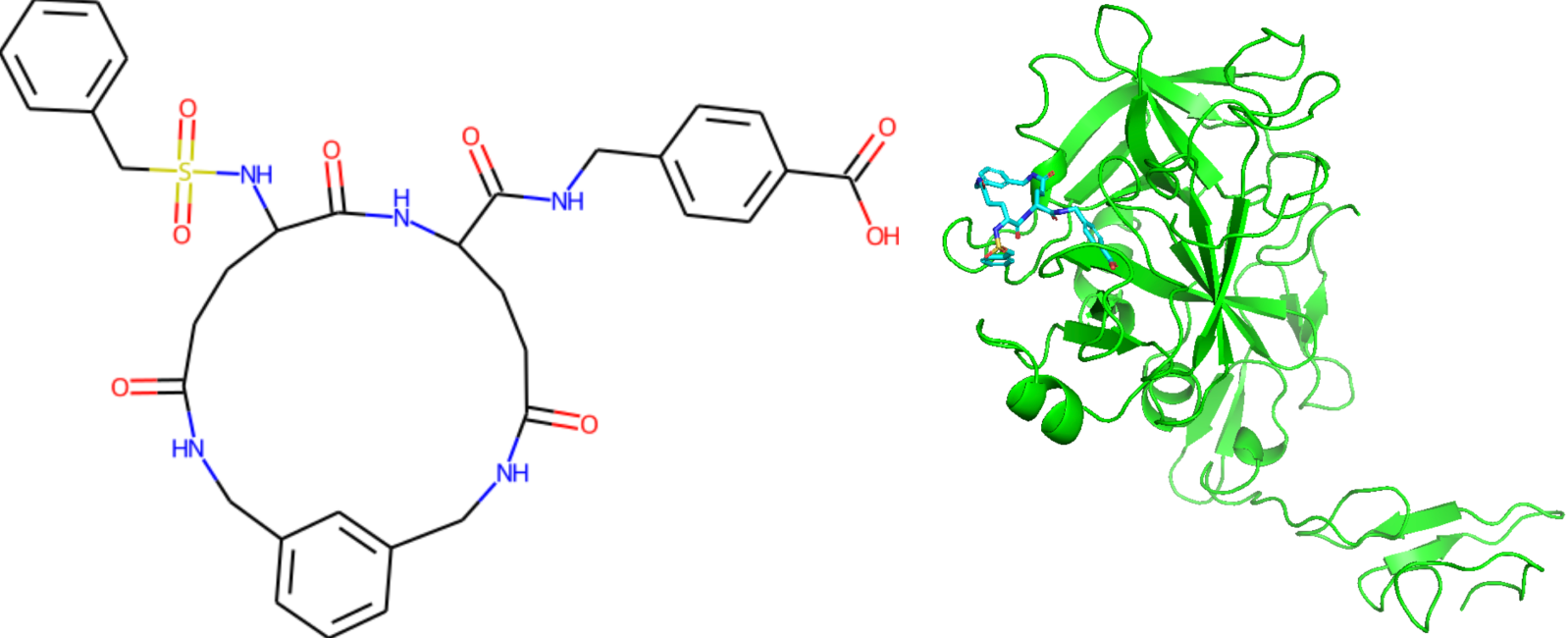
Wir verwendeten NVIDIAs BioNeMo, eine generative KI-Plattform für die Wirkstoffentdeckung, speziell dessen diffusionsgeneratives Modell DiffDock, um dieses Molekül an die aktivierte Form von Protein C anzudocken. Wir fanden heraus, dass die am besten bewertete Pose des Kandidatenmoleküls im aktiven Zentrum des Proteins liegt (siehe rechts). Wir nutzten auch MegaMolBART, um neue Moleküle für Protein-C-Inhibitorpaare und um bestimmte Inhibitoren herum zu generieren. Wir führten auch die Interpolation zwischen Paaren (100 Moleküle pro Paar) und die Hypersphärengenerierung um einige Inhibitoren herum durch (bis zu 60 Moleküle pro Inhibitor).
Ein Vergleich: Autoencoder vs. MegaMolBART
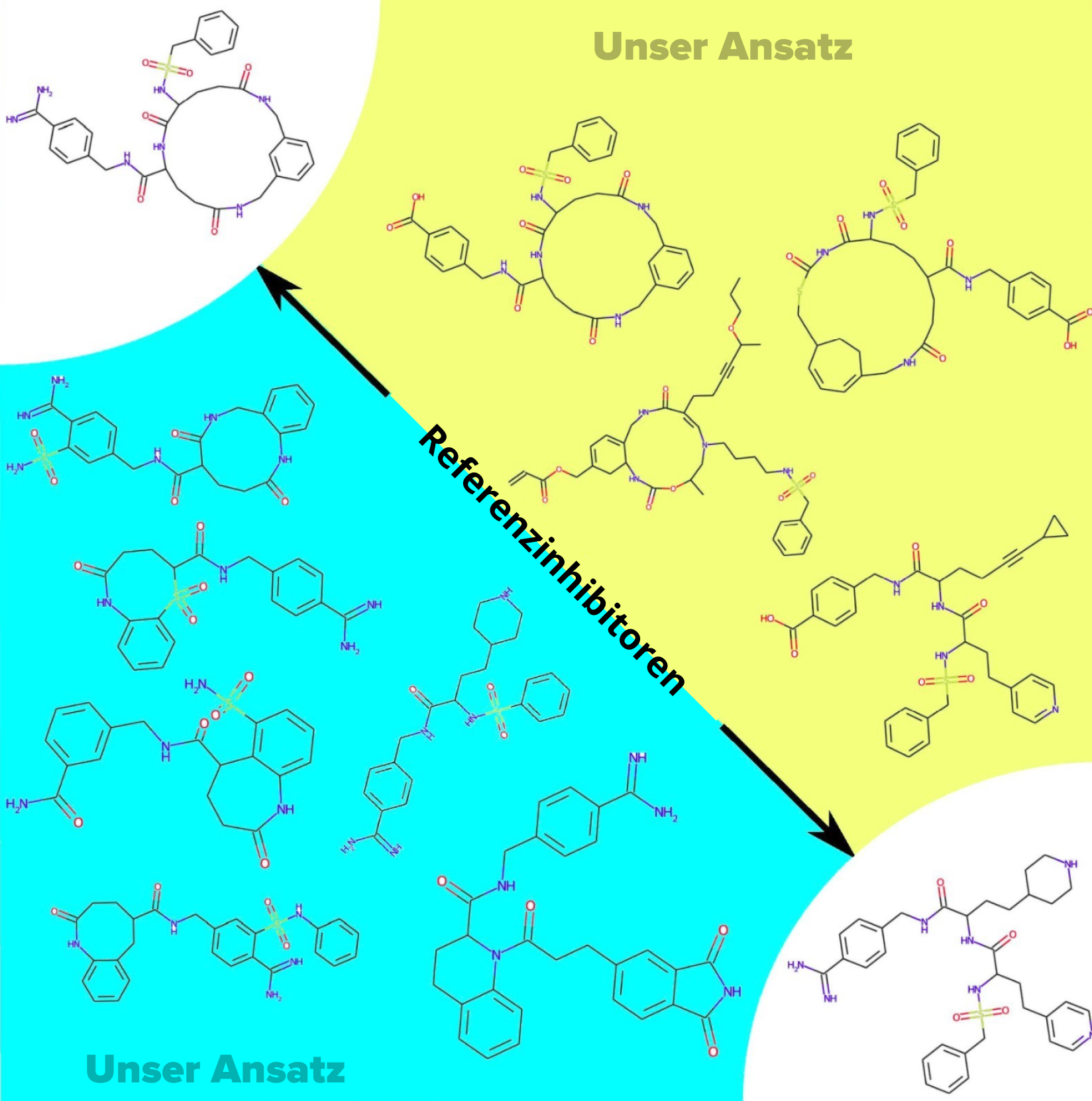
In dieser Arbeit wird ein Ansatz zur Identifizierung neuer Inhibitorkandidaten vorgestellt, der auf der Entwicklung eines Deep-Learning-Modells (Autoencoder) zur Generierung neuartiger Koagulans-Kandidaten basiert. Im Vergleich mit NVIDIAs MegaMolBART war unser Ansatz ebenfalls in der Lage, Kandidatenverbindungen auf der Grundlage leicht unterschiedlicher Prinzipien zu generieren. Darüber hinaus wurde einige Unterschiede zwischen unserer Methode und MegaMolBART sichtbar: MegaMolBART neigt dazu, mehr Moleküle zwischen nahen Inhibitoren zu generieren als unsere Methode. Zum Beispiel generierte es 14 Moleküle für ein Paar, während unsere Methode sechs produzierte.
Dieser Trend ist auch bei anderen Paaren zu beobachten. Unser Ansatz bewahrt die Referenzverbindungen besser. Andererseits erscheinen Cyclopropanringe (![]() ) und Cyclobutadienringe (
) und Cyclobutadienringe (![]() ) in unseren Ergebnissen, fehlen aber in den ursprünglichen Referenzinhibitoren. Unten sehen Sie einen Vergleich einer Interpolation zwischen zwei Protein-C-Inhibitoren aus unserem Ansatz. Wir sind fest davon überzeugt, dass der Erfolg beider generativer Ansätze dazu dient, mehr Optionen für die Generierung vorgeschlagener Kandidaten bereitzustellen und vielversprechend für die Zukunft der Wirkstoffentdeckung ist.
) in unseren Ergebnissen, fehlen aber in den ursprünglichen Referenzinhibitoren. Unten sehen Sie einen Vergleich einer Interpolation zwischen zwei Protein-C-Inhibitoren aus unserem Ansatz. Wir sind fest davon überzeugt, dass der Erfolg beider generativer Ansätze dazu dient, mehr Optionen für die Generierung vorgeschlagener Kandidaten bereitzustellen und vielversprechend für die Zukunft der Wirkstoffentdeckung ist.
Warum SoftServe
Bei SoftServe R&D können wir Ihnen helfen, ähnliche Pipelines für Projekte mit begrenzten Daten zu erstellen und diese mit unseren eigenen proprietären Daten zu optimieren. Unsere einzigartige Expertise und unsere Elite-Partnerschaft mit NVIDIA verschaffen uns Zugang zu den neuesten Werkzeugen auf diesem Gebiet.
Jetzt Kontakt aufnehmen