

Interactions between proteins are a fundamental part of many biological processes, with a significant portion of them involving peptides. Due to their chemical similarity to proteins, peptides have recently garnered significant attention as promising drug candidates. Consequently, the design of peptide-derived therapeutics heavily relies on understanding protein-peptide interactions.
Molecular docking is one of the most frequently used methods for predicting the preferred pose of one molecule (known as a ligand) relative to another (the receptor) when they form a stable complex. Methodologically, docking often consists of two stages: proposing a possible receptor-ligand pose and evaluating it using a scoring function.
SoftServe proposes an end-to-end molecular docking pipeline, illustrated schematically below.
The first stage involves a deep learning model that predicts the ligand’s docking spot on the receptor. A neural network considers atomic types, intramolecular bonds of both the receptor and ligand, and predicts a region near the receptor where a ligand is likely to attach during docking.
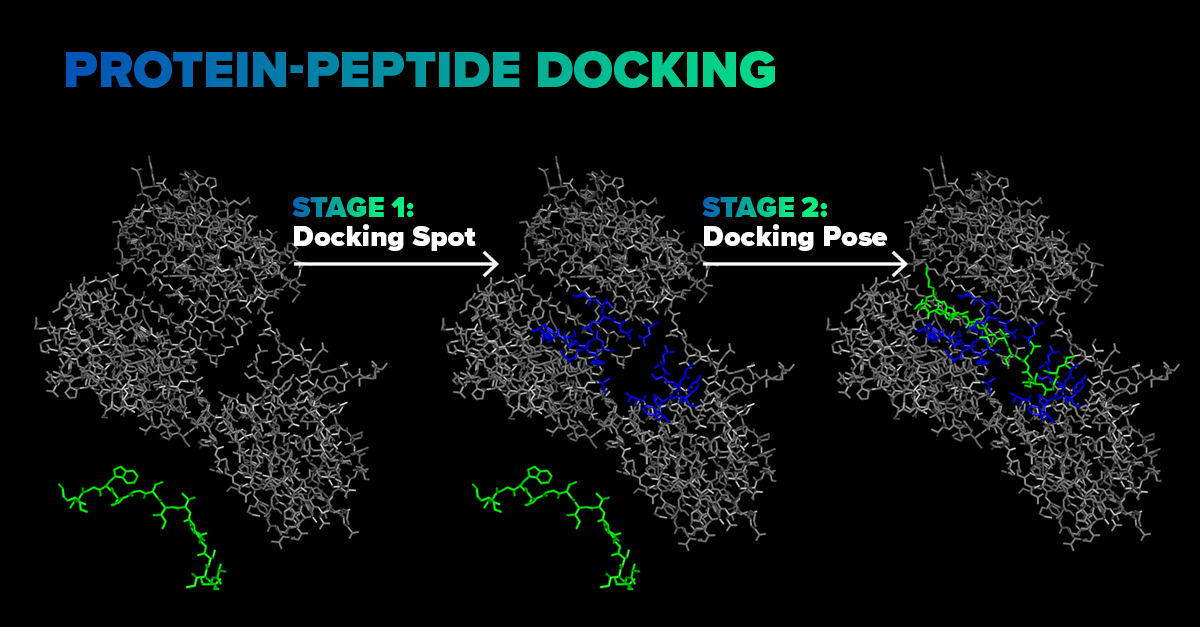
Such a prediction takes seconds and serves as a quick estimation to narrow the search space. We designed this model from scratch and trained it on protein-peptide complexes from the PepBDB dataset. The second stage is not yet fully automated; however, at this stage, the ligand’s position and conformation are fine-tuned. For this purpose, the ligand is automatically placed in front of the predicted docking spot, and DFTB+ — a Density Functional Theory (DFT)-based quantum mechanics simulation—is performed to determine the final ligand conformation and calculate the binding energy.
The developed end-to-end pipeline combines deep learning and DFT approaches to predict protein-peptide docking. It is worth noting that peptides are not the only class of possible ligands, as our solution can also dock other organic compounds. A key advantage of this approach is its two-stage setup, which allows for the localization of the docking spot in the first stage, significantly reducing calculation time and costs, while maintaining high precision through the use of DFT in the second stage.
Interested in advancing your research? SoftServe’s expertise in developing innovative R&D solutions, like our protein-peptide docking pipeline, helps tackle complex challenges in biotechnology and beyond.
Get in touch with our team to explore how we can support your next project.