

Ensembling Machine Learning Models to Boost Molecular Affinity Prediction
Initial stages of drug discovery require localization of factors causing a disease, understanding their molecular mechanism, then suggesting and testing drug leads. After a disease target has been identified, a list of drug candidates is drafted and screened for the target-candidate affinities, also known as a drug-target binding affinity (DTBA). The experimental methods quantifying DTBA are costly in terms of time and resources. Thus, computational approaches are needed to shrink the pool of tests by eliminating weakly scored candidates. Nowadays, machine learning (ML) techniques have become a powerful tool in the field of virtual screening.
The Art of ML Engineering
Typical ML-based study involves collection of task-specific data and selection of an ML technique. These techniques often rely on sequence or structural similarity of compounds, prompting for an effective parameterization of compounds to compare them. Since the task of drug-target interaction deals with drugs (ligands) and proteins (targets), it requires a parameterization of both. However, if there is one target in focus, only the ligands need to be parameterized. Multiple ways can be employed to parameterize chemical compounds, like, physico-chemical descriptors or fingerprint vectors, voxels (featurized 3D-cells) or molecular graphs, etc. This is the art of ML engineering - choosing the most effective data representation with the most appropriate ML technique to relate representations with labels - DTBAs, in our case.
The data availability is also a factor shaping the design of ML-based studies in this direction. Some public datasets provide a binary classification whether a particular protein-ligand pair binds or not (active/inactive), while other datasets include continuous data of, for example, inhibition constants Ki. Usually, the amount of binary data is larger than the amount of continuous data. Another issue is related to the distribution of continuous data - it is rather located within the active interval, being imbalanced towards the actively binding protein-ligands pairs.
Methods
In our recent paper we attempted to address the data issues and combine different ML approaches to compensate errors. We used six approaches to predict molecular DTBA of various ligands to human thrombin, however, the solution can be retrained for other proteins, as well. In particular, Support Vector Machine (SVM), Random Forest (RF), Catboost, feed-forward neural network (FFNN), graph neural network (GNN), and Bidirectional Encoder Representations from Transformers (BERT) were employed. As one receptor (human thrombin) is considered, only ligands need to be parameterized. The first four approaches - SVM, RF, CatBoost, FFNN - use molecular fingerprints of ligands. The fifth approach - GNN - uses graph representations by treating atoms and bonds as graph nodes and edges. BERT works directly with string representations of ligands. None of the mentioned representations use spatial coordinates, which was one of our essential requirements to predict DTBAs for novel ligands with unknown conformations. All models are wrapped into a pipeline shown in the figure below.
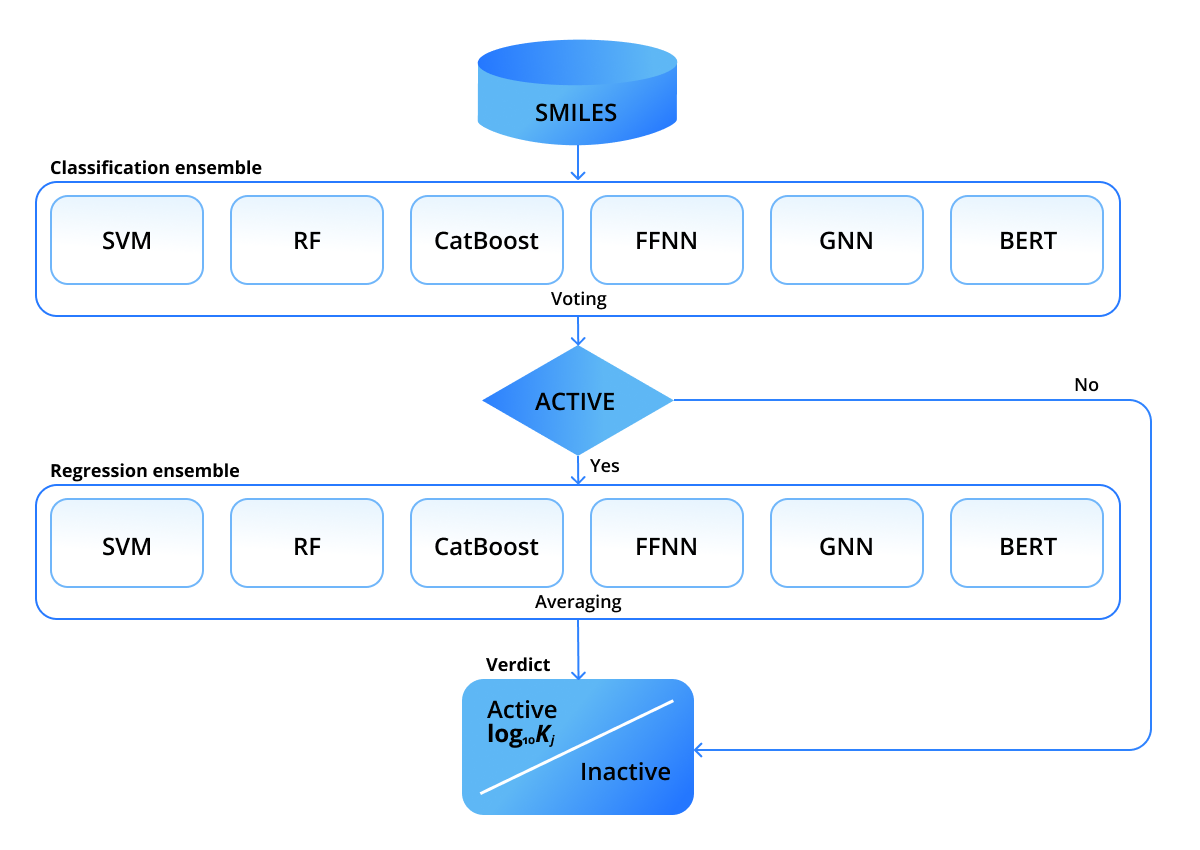
It comprises two stages: the first stage decides whether a ligand is active or inactive, then, if it is classified as active, the second stage predicts its inhibition constant Ki. The predictions within the stages are combined in an ensemble-like manner. We employed a rather strict voting rule within the classification ensemble: a ligand is classified active only if ALL the models classify it as active. Such an active-demanding rule is dictated by industry needs: only potentially active ligands are further passed to wet-lab experiments. The prediction of the regression ensemble is calculated as an average over predictions of individual models.
The two-ensemble setup allowed us to make use of all available data, using both binary (active/inactive) labels and continuous inhibition constants. Each of the methods yielded two models trained on the two arrays of data - classification and regression. The modular structure within the ensembles allows one to various ensembling scenarios.
Data
A number of public datasets provide data for inhibition constants Ki, as well as for a classification to active/inactive ligands towards different receptors. The lower Ki is, the stronger (more active) inhibitor is, and vice versa. There is no strict threshold for the inhibition constant between active and inactive ligands, however, Ki of 10 000 nM is often considered as such a delimiter.
As our pipeline consists of the classification and regression ensembles, we prepared two datasets to train the corresponding models. We collected ~30 000 ligand samples of two activity classes with respect to human thrombin which constituted our classification dataset. The dataset for regression consisted of ~4000 samples with corresponding values of inhibition constants within the range of [0:30 000 nM]. The exceed of the Ki range over 10 000 nM is aimed to allow the regression models to learn the inhibition constants for very weak inhibitors. In concentration measurements, the error increases proportionally to the concentration itself, therefore, it is convenient to work in terms of decimal logarithm of inhibition constants log10Ki to balance error contributions at different concentration ranges.
Results
First, we show the performance of the classification models with a k-fold validation. The dataset was split into five equal-sized non-intersecting folds: every model was retrained five times, each time having a different fold as test subset, while the remaining four folds served as train subset. The averaged (over folds) precision values are presented in the table below.
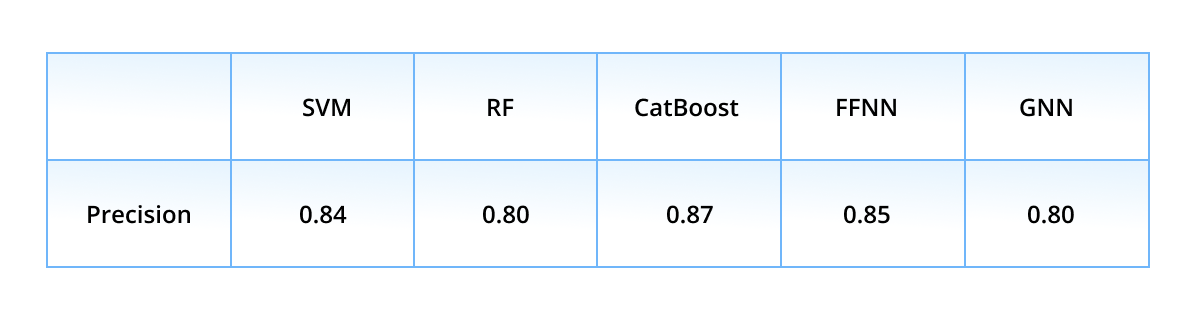
Precision is the most important score here out of other ones, because it relies on true and false actives - to remind the reader, only active ligands (either true or false predicted) are passed to wet-lab experiments. Thus, the “leaders” in the precision race are CatBoost (0.87), FFNN (0.85), and SVM (0.84), while the ensembled precision score is even higher - 0.95. Such a high result is achieved due to the strict voting rule - the ensemble classifies a ligand as active only if all models classify it as active. Different ML algorithms are prone to different classification errors, but mutually exclude the errors of their mates.
Next, we discuss the performance on the regression task. The table below presents the mean squared error MSE and coefficient of determination R2 averaged over five validation folds.
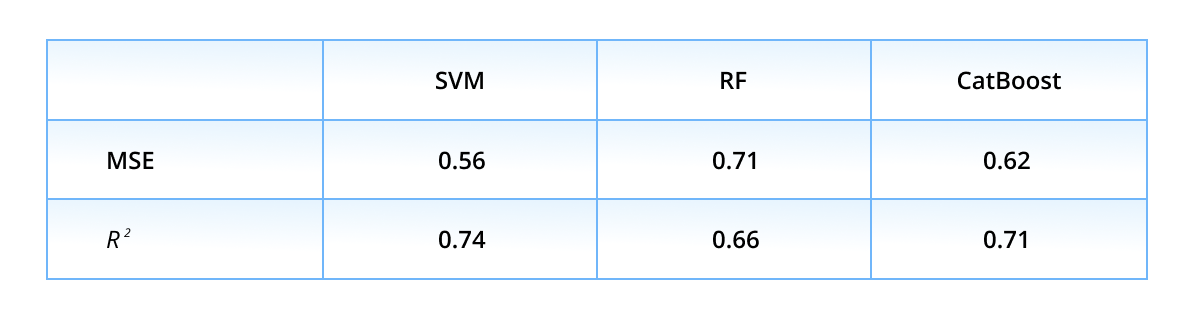
One can see, the list of the top-performers differs from the classification list: the lowest MSE and the highest R2 scores are achieved with the SVM approach, followed by CatBoost, GNN, and FFNN. The average over predictions of six models constitutes the ensembled prediction, which notably improves in comparison to individual ones.
Concluding remarks
Typical tasks of high-throughput screening might require billions of in silico experiments, promoting the family of machine learning methods, as they are faster than traditional approaches. We suggest an ML-based pipeline to predict binding affinities between small organic molecules and proteins. The pipeline unites two subsequent ensembles -- classification and regression. With classification followed by regression, not only drug candidate class (active/inactive) is assessed, but also the strength of association, which is of high interest for the pharma industry because it allows for selection of candidates with the highest activities.
The two-ensemble pipeline makes use of all available affinity data, reducing the error-rate and being fast enough for the high-throughput screening. The diversity of the methods allowed us to exclude (on the classification) or compensate (on the regression) the errors made by their ensemble mates. Although the performance of the discussed pipeline is validated in the case of human thrombin, it can be applied for other proteins, as well.
Access the full study at ScienceDirect, “Ensembling Machine Learning Models to Boost Molecular Affinity Prediction.”
Read more about the topic: “Protein-Peptide Docking.”
Let's talk about how an R&D-focused extended team can bring you value, and support you on your performance optimization journey.